Introduction
Overview
Teaching: 10 min
Exercises: 15 minQuestions
What parallel computing is and why it is important?
How does a parallel program work?
Objectives
Explain differences between a sequential and a parallel program
Introduce types of parallelism available in modern computers
Parallel computing is the science of solving problems using multiple computer processors at the same time.
Why parallel computing?
-
From the beginning of electronic computers to about 2010 or so, computer engineers were able to build faster and faster CPUs. Clock speeds went from about 60 MHz around 5 GHz between about 1990 and 2010. Programs could be sped up just by buying the latest chip every year.
-
This growth has effectively ended due to limitations of miniaturization, power consumption, and heat dissipation.
-
It is increasingly expensive to make a single processor faster. Using a larger number of moderately fast commodity processors to achieve the same (or better) performance is more practical.
Example applications
Here are just a few examples of how parallel computing is helping to accelerate research and solve new problems.
- Numeric weather prediction
- Combining current observations and mathematical models to forecast the future state of the weather
- Seismic data processing
- Analyze seismic data to find oil and gas-bearing layers
- Computational fluid dynamics; finite element modelling
- tidal wave simulation; design of automobiles, ships, aircraft
- Commerce
- credit scoring, risk modelling, fraud detection, all GPU-accelerated
- Pharmaceutical design
- simulations of molecular dynamics
- Train AI models
- Large Language Models, Generative AI, robotics
Benefits of parallel computing:
- Compared to sequential (also called “serial”) computing, the number of problems you can solve practically with parallel computing is huge.
- It allows us to solve problems in a shorter time.
- Allows solving larger, more detailed problems.
- Parallel computing is better suited for modelling, simulating, and understanding complex, real-world phenomena.
- Handle high-velocity data streams.
Limitations and disadvantages:
- Some problems can not be broken into pieces of work that can be done independently
- Not every problem can be solved in less time with multiple compute resources than with a single computing resource. Some problems may not have enough inherent parallelism to exploit.
- Large-scale parallel computing equipment is expensive.
- Developing parallel code is not easy.
- It requires expertise, time, and effort.
Before you start your parallel computing project
- Select the most appropriate parallelization options for your problem.
- Estimate the effort and potential benefits.
In this course, we hope to give you the knowledge to make the right decisions on parallel computing projects.
How does a parallel program work?
Let us start by exploring how a sequential (non-parallel) program works.
Traditional sequential program

- A problem is broken into a set of instructions
- Instructions are executed in a specified order on a single CPU
- Only one instruction can be executed at a time
Parallel program
In parallel computing, individual instructions execute concurrently on different CPUs and/or computers.

- A problem is broken into parts that can be done at the same time
- Each part is further broken down into a set of instructions
- Instructions from each part execute simultaneously on different processors
- Requires an overall control & coordination mechanism.
Discussion exercise
Pretend it’s the days before electronic computers, and you have been assigned the task of computing the sum of 1,000 four-digit numbers as rapidly as possible. You hold in your hands a stack of 1,000 index cards, each containing a single number, and you are in charge of 1,000 accountants, each with a pencil. You may choose to use the services of any number of these accountants. The accountants are sitting at desks in a cavernous room. The desks are organized into 25 rows and 40 columns. Each accountant can pass cards to the four accountants nearest them— in front, in back, to the left, and to the right.
Discuss with your classmates:
- How many accountants should we use?
- How will we get the cards to right accountants?
- How will we turn the subtotals generated by the active accountants into a grand total?
- What should we do differently if there were 10,000 cards (but still 1,000 accountants)?
You do not need to arrive at a perfect plan. (Indeed, you don’t have enough information to devise a perfect plan!) But try to sketch a rough plan, figure out some ideas, talk about whether those ideas have merit or not, and think what other information you would want to carry this out this imaginary task.
(Adapted from Michael J. Quinn, “Parallel Programming in C with MPI and OpenMP”)
Key Points
Parallel computing is much better suited for modelling, simulating and understanding complex, real-world phenomena.
Modern computers have several levels of parallelism
Parallel Computer Architecture
Overview
Teaching: 15 min
Exercises: 0 minQuestions
How is a typical CPU organized?
How are parallel computers organized?
Objectives
To make full use of computing resources the programmer needs to have a working knowledge of the parallel programming concepts and tools available for writing parallel applications. To build a basic understanding of how parallel computing works, let’s start with an explanation of the components that comprise computers and their function.
Von Neumann’s Architecture
John von Neumann published a general description of a computer architecture in 1945. It describes the stored-program principle where computer memory is used to store both instructions and data. The von Neumann architecture is still an excellent model for the computer processors we use today.

- Central Processing Unit (CPU):
- The electronic circuit responsible for executing the instructions of a computer program.
- The CPU contains:
- Control unit:
- Reads and interprets instructions from the memory unit.
- Controls the operation of the ALU, memory and input/output devices, telling them how to respond to the program instructions.
- Arithmetic Logic Unit (ALU):
- Carries out arithmetic (add, subtract etc) and logic (AND, OR, NOT etc) operations.
- Registers
- Registers are high-speed storage areas in the CPU. All data must be stored in registers before they can be processed by the ALU.
- Memory Unit:
- Consists of random access memory (RAM) modules. RAM is slower than registers but much faster than a hard drive (secondary memory), and directly addressable by the CPU.
- Input/Output Devices:
- The interface to the outside world, e.g. hard drives, network, keyboard, mouse, monitor, etc.
Parallel computers still follow this basic design, just multiplied in units. A physical CPU chip in the 2020s may contain several von Neumann CPUs, which we usually call cores when we need to make the distinction. An individual computer might also contain more than one of these multi-core CPU chips. But essential layout of a modern core is the same as the von Neumann architecture shown above.
For the remainder of this course we may sometimes say “CPU” when we should more precisely say “core”, because the physical CPU chip is not the important component when thinking about parallel programming. The core executes instructions in sequential order, not in parallel. The core is the basic building block of parallel computing hardware.
Classifications of parallel computers
There are various academic classifications of parallel computer architectures. Here are two you can look up later if you’re interested:
- Flynn’s taxonomy (1966) is based on whether there are one or more instruction streams and one or more data streams.
- Feng’s classification (1972) is based on the number of bits than can be processed at one time.
Flynn’s taxonomy is probably the most widely known. It gives rise to terms like “Single-Instruction, Multiple-Data” (SIMD) and “Multiple-Instruction, Multiple-Data” (MIMD).
Memory organization
We can see from the von Neumann architecture that a processing core needs to get both data and instructions from memory, and write data back to that same memory. Engineering constraints mean that the processor can typically carry out instructions far faster than data can be read and written to main memory, so how memory is organized has important consequences for the design and execution of parallel programs.
There are currently two main ways to organize memory for parallel computing:
- Shared memory
- Distributed memory
A hybrid of these two could be considered a third way.
Shared memory
- Multiple processors can operate independently but share the same memory resources.
- All processors have equal access to data and instructions in this memory
- Changes in a memory done by one processor are visible to all other processors.
Shared memory computers can be further sub-divided into two categories:
- Uniform Memory Access (UMA)
- Non-Uniform Memory Access (NUMA)

- All processors share memory uniformly, i.e. access time to a memory location is independent on which of the processors.
- When many CPUs are trying to access the same memory they can be “starved” of data because only one processor can access the computer’s memory at a time.
- Doesn’t scale well to large numbers of processors.

- NUMA has separate memory for each processor.
- CPUs are physically linked using a fast interconnect.
- A CPU can access its local memory faster than non-local memory (memory local to another processor or memory shared between processors).
- Most present-day machines are NUMA.
In a shared memory system it is only necessary to build a data structure in memory and pass references to the data structure to parallel subroutines. For example, a matrix multiplication routine that breaks matrices into a set of blocks only needs to pass the indices of each block to the parallel subroutines.
Advantages
- User-friendly programming.
- Fast data sharing due to a close connection between CPUs and memory
Disadvantages
- Not highly scalable. Adding more CPUs increases traffic on the shared memory-CPU path.
- Lack of data coherence. The change in the memory made by one CPU needs to be reflected to the other processors, otherwise, the different processors will be working with inconsistent data.
- The programmer is responsible for synchronization.
Distributed memory

- In a distributed memory system each processor has a local memory that is not accessible from any other processor
- Programs on each processor interact with each other using some form of a network interconnect (Ethernet, Infiniband, Omni-Path, etc.).
- A distributed memory program must create copies of shared data in each local memory. These copies are created by sending a message containing the data to another processor.
Advantages
- Each processor can use its local memory without interference from other processors.
- There is no inherent limit to the number of processors; the size of the system is constrained only by the network used to connect processors.
Disadvantages
- Complexity of programming: The programmer is responsible for many of the details associated with data communication between processors.
- It may be difficult to map complex data structures from global memory, to distributed memory organization.
- Longer data access times, if the data is not local to the process.
Hybrid Distributed-Shared Memory
 Practically all HPC computer systems today employ both shared and distributed memory architectures.
Practically all HPC computer systems today employ both shared and distributed memory architectures.
- The shared memory component can be a shared memory machine and/or graphics processing units (GPU).
- The distributed memory component is the networking of multiple shared memory/GPU machines.
The important advantage is increased scalability. Increased complexity of programming is an important disadvantage.
Programming models are memory models
When designing a parallel solution to your problem, the major decision is usually “which memory model should we follow?”
-
A shared-memory solution can only be scaled up as far as the largest shared-memory machine available to you.
-
In a distributed-memory solution, data must be copied from one node to another via messages, and this communication overhead can reduce efficiency to the point where scaling-up produces no benefit.
You’ll learn more about these two major models in the OpenMP programming and MPI programming segments of this Summer School.
Counting CPUs
Imagine you’re looking at a product description from a computer vendor. You’ve determined that the computer has two CPU slots, and that each slot holds a 4-core Intel i7-6700 processor. How many “von Neumann CPUs” does this machine have?
Solution
The machine has 8 von Neumann CPUs, or more briefly, 8 cores.
The term “CPU” these days has two distinct meanings, depending on context. A hardware CPU is a physical object (e.g. an i7-6700) that will usually have several “cores”. A “core” corresponds to the “von Neumann CPU” we described earlier in this lesson. Consequently, when reading about software and parallel programming, you may see the term “CPU” used where the term “core” would be more correct.
If you followed the link to the chip description, you might have seem the term “hyperthreading”, which further confuses the matter. Hyperthreading is a proprietary Intel technology that uses software to try to double the processing capacity of each core. This works well for many low-intensity workloads, but for high-intensity parallel computing, performance is usually limited by the number of physical cores. So we don’t count the extra “logical cores” supposely provided by hyperthreading.
Key Points
Sequential computers, including modern CPU cores, resemble very well von Neumann’s 1945 design.
Parallel computers can be characterized as collections of von Neumann CPUs.
Parallel computers may be shared-memory, distributed-memory, or both.
Parallel Programming Models
Overview
Teaching: 15 min
Exercises: 10 minQuestions
What levels of parallelism are available in modern computer systems?
Objectives
Terms: instruction, thread, process
Parallel programming models: vectorization, multithreading, message passing
Parallel Programming Models
Developing a parallel application requires an understanding of the potential amount of parallelism in your application and the best ways to expose this parallelism to the underlying computer architecture. Current hardware and programming languages give many different options to parallelize your application. Some of these options can be combined to yield even greater efficiency and speedup.
Levels of Parallelism
There are several layers of parallel computing:
- Vectorization (processing several data units at a time)
- Multithreading (a way to spawn multiple cooperating threads sharing the same memory and working together on a larger task). Multithreading provides a relatively simple opportunity to achieve improved application performance with multi-core processors.
- Process-based parallelism
Available hardware features influence parallel strategies.
Example workflow of a parallel application in the 2D domain.
- Discretize the problem into smaller cells or elements
- Define operations to conduct on each element of the computational mesh
- Stencil operation involves a pattern of adjacent cells to calculate the new value for each cell.

An example of a stencil operation is blurring an image: Information from neighboring pixels is introduced into each pixel.
\(x_{i,j}^{new}=x_{i-1,j}+x_{i,j-1}+x_{i,j}^{old}+x_{i+1,j}+x_{i,j+1}\)
- Repeat computations for each element of the mesh
Vectorization
Vectorization can be used to work on more than one unit of data at a time.

- Many modern CPUs can operate on several pieces of identical data (int, float, double) at one time.
- An example are CPUs which support AVX instructions.
- The vector operation shown in the figure is conducted on eight floats.
- The following code shows how to set eight values (twice) and then subtract them using single AVX instructions.
Example AVX2 program
avx2_example.c
#include <immintrin.h>
#include <stdio.h>
int main() {
/* Initialize the two argument vectors */
__m256 evens = _mm256_set_ps(2.0, 4.0, 6.0, 8.0, 10.0, 12.0, 14.0, 16.0);
__m256 odds = _mm256_set_ps(1.0, 3.0, 5.0, 7.0, 9.0, 11.0, 13.0, 15.0);
/* Compute the difference between the two vectors */
__m256 result = _mm256_sub_ps(evens, odds);
/* Display the elements of the result vector */
float *f = (float *)&result;
printf("%f %f %f %f %f %f %f %f\n",
f[0], f[1], f[2], f[3], f[4], f[5], f[6], f[7]);
return 0;
}
Compiling: gcc -mavx avx2_example.c -o avx2_example
- Typically we would not call these functions explicitly in application code. We would typically just write a loop and let the compiler choose what CPU-specific functions to call.
Threads
Most of today’s CPUs have several processing cores. We can use threading to engage several cores to operate simultaneously on four rows at a time as shown in the next figure.

Technically, a thread is defined as an independent stream of instructions that can be scheduled to run by the operating system. From a developer’s point of view, a thread is a piece of code that can execute concurrently with other threads, but that may share certain things like access to the same memory.
On Linux systems (and many others), every process has at least one thread, but may have more. A thread is also called “light-weight process” or LWP.
A “multi-threaded” program is one which is designed to run more than one thread to take advantage of a shared-memory or multi-core architecture.
POSIX Threads (Pthreads) and OpenMP represent the two most popular implementations of this model. The Pthreads library is a low-level API providing extensive control over threading operations. It is very flexible and provides very fine-grained control over multi-threading operations. Being low-level it requires multiple steps to perform simple threading tasks.
OpenMP is a much higher level and much simpler to use.
Below is some sample code in C which uses OpenMP.
The line #pragma omp parallel for tells an OpenMP-compliant compiler
that the following loop should be executed in multiple threads.
Example OpenMP program
omp_example.c
#include <omp.h>
#include <stdio.h>
int main()
{
int i;
int n=10;
float a[n];
float b[n];
for (i = 0; i < n; i++)
a[i] = i;
#pragma omp parallel for
/* i is private by default */
for (i = 1; i < n; i++)
{
b[i] = (a[i] + a[i-1]) / 2.0;
}
for (i = 1; i < n; i++)
printf("%f ", b[i]);
printf("\n");
return(0);
}
- Compiling:
gcc -fopenmp omp_example.c -o omp_example - Running:
OMP_NUM_THREADS=2 ./omp_example
Distributed memory / Message Passing
Further parallelization can be achieved by spreading out computations to a set of processes (tasks) that use their own local memory during computation.
Multiple tasks can reside on the same physical machine and/or across an arbitrary number of machines.
Tasks exchange data through communications by sending and receiving messages.
MPI is the dominant standard for message-passing programming, and OpenMPI and Intel MPI are two popular implementations of that standard. Here is some sample code which does the MPI equivalent of “Hello, world!”

MPI example
mpi_example.c
#include <mpi.h>
#include <stdio.h>
int main(int argc, char** argv) {
// Initialize the MPI environment
MPI_Init(NULL, NULL);
// Get the number of processes
int world_size;
MPI_Comm_size(MPI_COMM_WORLD, &world_size);
// Get the rank of the process
int world_rank;
MPI_Comm_rank(MPI_COMM_WORLD, &world_rank);
// Print off a hello world message
printf("Hello world from rank %d out of %d Processors\n", world_rank, world_size);
// Finalize the MPI environment.
MPI_Finalize();
}
- Compiling:
mpicc mpi_example.c -o mpi_exampleorgcc mpi_example.c -lmpi -o mpi_example -
Running:
srun -n 4 ./mpi_example - Passing messages takes time
Resources:
- Crunching numbers with AVX and AVX2
- ACENET OpenMP course materials
- More OpenMP learning resources
- ACENET MPI course materials
- ACENET course schedule and sign-ups
Key Points
There are many layers of parallelism in modern computer systems
An application can implement any or all of vectorization, multithreading, and message passing
Performance and Scalability
Overview
Teaching: 30 min
Exercises: 30 minQuestions
How do we measure parallel performance?
Objectives
Learn how to measure parallel scaling.
Understand limits to parallel speedup.
Although speed is not the only reason one might be interested in parallel programming, speed is still a central consideration. If you want to solve larger problems using parallel computing, it won’t do if the parallel code runs slower than the sequential code.
Therefore it’s important to be able to measure and analyze performance.
Speedup
Parallel speedup is defined as the ratio of the runtime of the best sequential algorithm (on one processor) to the runtime of the parallel algorithm to solve the same problem on $N$ processors:
[Speedup = \frac{T_s}{T_p}]
…where $T_s$ is sequential runtime and $T_p$ is parallel runtime. Sequential runtime is (usually!) longer than parallel runtime, so a larger speedup is better.
Optimally, the speedup from parallelization would be linear. Doubling the number of processing elements should halve the runtime, and doubling it a second time should again halve the runtime. That would mean that every processor would be contributing 100% of its computational power. However, very few parallel algorithms achieve optimal speedup. Most of them have a near-linear speedup for small numbers of processing elements, which flattens out into a constant value for large numbers of processing elements.
Efficiency
Efficiency is the ratio between the actual speedup and the ideal speedup obtained when using a certain number of processors. We just argued that the ideal speedup is proportional to the number of processors, $N$, so:
[Efficiency=\frac{Speedup}{N}=\frac{T_s}{T_p*N}]
Efficiency can be also understood as the fraction of time for which the processors are doing useful work, or the fraction of the processors which are doing useful work on average.
- 0-100%
- Depends on the number of processors
- Varies with the size of the problem
When writing a parallel application we want processors to be used efficiently.
Amdahl’s law
We can think of a program or algorithm as having parallelizable and non-parallelizable parts. Parallelizable parts are those segments of the code that can execute on separate processors doing separate work. Non-parallelizable parts are those segments of the code which can only be done by one process, or equivalently, parts which do duplicate work if executed by more than one process.
Reading in a parameter which every process must have, for example, is a non-parallelizable operation. You can either have one process read, or you can have all processes read, but since all processes would be doing exactly the same work, no speed advantage would be gained by parallelizing the operation. Therefore input is a non-parallelizable part of most programs.
Amdahl’s Law takes this division of code into parallelizable and non-parallelizable parts to describe the maximum speedup of an algorithm.
Express the time to run the sequential code as the sum of the time to run the parallelizable fraction, $P$, and the time to run the non-parallelizable fraction, $S$ (for sequential):
[T_s = T_s S + T_s P; S+P=1]
The time to run the parallel code is reduced by the number of processors $N$, but only for the parallelizable part. Furthermore, the parallel program may have to do certain operations that the sequential code does not. These operations might include, for example, setting up communications with other processes, or waiting for other processes to complete so that shared memory is consistent. We encapsulate these extra operations as parallel overhead, $K$:
[T_p = T_s S + T_s \frac{P}{N} + K]
If we make the extremely optimistic assumption that $K$ is negligibly small and substitute the above into the definition of speedup, we get:
[Speedup = \frac{T_s}{T_p} = \frac{1}{S+\frac{P}{N}}]
This equation is known as Amdahl’s law. It states that the speedup of a program from parallelization is limited by the fraction of the program that can be parallelized.
For example, if 50% of the program can be parallelized, the maximum speedup using parallel computing would be 2 no matter how many processors are used.
| Speed Up | Efficiency |
|---|---|
 |
 |
Amdahl’s law highlights that no matter how fast we make the parallel part of the code, we will always be limited by the sequential portion. Furthermore, if the parallel overhead $K$ is not actually small, that’s even worse news. It is entirely possible for the run time of a parallel program to be worse than the run time of the equivalent sequential program due to $K$.
This suggests that parallel computing is only useful when the number of processors is small, or when the problem is perfectly parallel, i.e., embarrassingly parallel. Amdahl’s law is a major obstacle in boosting parallel performance.
But notice that Amdahl’s law assumes that the total amount of work is independent of the number of processors, that is, the problem is the same size no matter how many processors we use. This type of scaling is referred to as Strong Scaling.

Gustafson’s law
It is very common, however, that we have some control over the size of our problem, and we are using parallel computing to handle larger problems than we can in sequential. For example, we might be doing some simulation on a grid where we can choose how fine to make the grid. Increasing the number of points on the grid increases (we hope) the precision of our results, but also requires more computing.
This scenario is called Weak Scaling:

If we can arrange that the total amount of work to be done in parallel varies linearly with the number of processors, speedup will be given by Gustafson’s law, shown here without derivation:
[\large{Speedup = N − S * (N − 1)}]
where $N$ is the number of processors and $S$ is the sequential fraction as before. (We also continue to assume that the parallel overhead $K$ is negligible.)
| Speed Up | Efficiency |
|---|---|
 |
 |
The theoretical speedup is more optimistic in this case. We can see that any sufficiently large problem can be solved in the same amount of time by using more processors. We can use larger systems with more processors to solve larger problems.
Example problem with weak scaling.
Imagine that you are working on a numeric weather forecast for some country. To predict the weather, you need to divide the whole area into many small cells and run the simulation. Let’s imagine that you divided the whole area into 10,000 cells with a size of 50x50 km and simulation of one week forecast took 1 day on 1 CPU. You want to improve forecast accuracy to 10 km. In this case, you would have 25 times more cells. The volume of computations would increase proportionally and you would need 25 days to complete forecast. This is unacceptable and you increase the number of CPUs to 25 hoping to complete the job in 1 day. Gustafson’s Law says you have some reasonable hope of it working.
Example problem with strong scaling.
Imagine that you want to analyze customer transactional data for 2019. You cannot add more transactions to your analysis because there was no more. This is fixed-size problem and it will follow strong scaling (Amdahl’s) law.
Any real-life problem falls in one of these 2 categories, although it is not always practical to maintain a precise linear proportion between problem size and number of processors. The type of scaling is a property of a problem that cannot be changed, but understanding what kind of scaling to expect helps us make sensible choices about the number of processing elements so that we use them efficiently.
Measuring Parallel Scaling
It is important to measure the parallel scaling of your problem before running long production jobs.
- To test for strong scaling we measure how the wall time of the job scales with the number of processing elements (openMP threads or MPI processes).
- To test for weak scaling we increase both the job size and the number of processing elements.
- The results from these tests allow us to determine the optimal amount of CPUs for a job.
Example: Generating an image of a Julia set
The example program julia_set_openmp.c calculates an image of a Julia set. It is adapted from one written by John Burkardt and released under the GNU LGPL license.

The program finds a set of points in a 2D rectangular domain with width W and height H that are associated with Julia set.
The idea behind a Julia set is to choose a complex constant $c$, and then for each point $z_0$ in a complex domain, repeatedly evaluate this expression:
[z_{n+1}=z_{n}^2+c]
If the sequence of values $z_{n}$ remains bounded, the initial $z_0$ is in the set; if the sequence diverges, the initial $z_0$ was outside the set. For each complex constant $c$ one gets a different Julia set.
To construct the image, we take rectangular region of pixels (x,y) and map each pixel to a complex number: $z_0=x+iy$. Each pixel then represents a starting point $z_0$ for the series $z_{n}$. We start computing the series, and if $\lvert z \lvert$ grows past some large number (1000 in this implementation) we conclude it’s outside the set and color it white. If it doesn’t diverge in a reasonable number of steps (200 in this implementation), we conclude it’s inside the set and draw it in red.
The program takes the width and height of the image (in pixels) and the number of parallel threads as arguments. It will print the number of threads and computation time on screen. It’s a good idea to repeat each computation several times to observe how much variance there is.
Running strong and weak scaling tests
We can run this sample program in both strong and weak scaling modes.
To test the strong scaling run the program with fixed size (width=2000, height=2000) and different numbers of threads (1-16).
To measure weak scaling we run the code with different numbers of threads and with a correspondingly scaled width and height.
Once the runs are completed we fit the strong and weak scaling results with Amdahl’s and Gustafson’s equations to obtain the ratio of the sequential part (s) and the parallel part (p).
Compiling and Running the Example
- Connect to a cluster (if you haven’t already), with X11 forwarding enabled so that you’ll be able to display some graphs.
- If you’re on Windows using MobaXterm, check that the “X” icon in the upper right-hand corner is colored.
- If you on a Mac, start XQuartz, then start a terminal and log in with
ssh -X username@cluster...The uppercase-Xturns on X11 forwarding.
- Download and unpack the code:
wget https://acenet-arc.github.io/ACENET_Summer_School_General/code/julia_set.tar tar -xf julia_set.tar cd scaling - Compile the program
julia_set_openmp.cgcc -fopenmp julia_set_openmp.c - Run on 2 CPUs (for example)
./a.out 200 200 2JULIA_OPENMP: C/OpenMP version. Plot a version of the Julia set for Z(k+1)=Z(k)^2-0.8+0.156i Height, width, threads, seconds: 200, 200, 2, 0.0103905 TGA_WRITE: Graphics data saved as 'julia_openmp.tga' JULIA_OPENMP: Normal end of execution.The program generates image file
julia_openmp.tga, which you may be able to see usingdisplay julia_openmp.tga. (You might have to turn on X11 graphics first if you get the errordisplay: unable to open X server) - To measure strong scaling submit array job:
sbatch submit_strong.sh#!/bin/bash #SBATCH --cpus-per-task=16 #SBATCH --time=5 #SBATCH --mem-per-cpu=1000M for nthreads in $(seq 1 16) do for replicate in 1 2 3 do ./a.out 200 200 $nthreads done done - Extract the scaling data from the raw output.
The data we need are in the
slurm-1234567.outfile. (Make sure the job ID matches the job you just submitted; you might do this more than once!) We need the thread counts and the run times, which you can extract with a utility likegreporawk:awk '/threads, seconds:/ {print $7, $8}' slurm-1234567.out > strong-1234567.csv - Fit the data with Amdahl’s law.
The python script will plot the data, and try to fit Amdahl’s law to the data
by finding a sequential fraction $S$ that fits best.
(To see the graph you may need to have an X11 server running and X11 forwarding enabled.)
module load python scipy-stack python strong_scaling.py strong-1234567.csvThe Python script will also save the figure as “strong_scaling.svg”, and you can re-display it using
display strong_scaling.svg.
Testing weak scaling
Find the
submit_weak.shjob script in thescalingdirectory you recently unpacked. It’s incomplete: Replace the....s with suitable numbers to evaluate Gustafson’s Law (the weak scaling law) like we just evaluated Amdahl’s law. What is the problem size in this example? How should it relate to the number of threads?Submit the job. When it has run, extract the data as we did above and use the
weak_scaling.pyscript to plot the data and a best-fit line under the assumptions of Gustafson’s law.Are your graphs smooth? If not, why do you suppose that might be?
Compare the speedups obtained using strong and weak scaling tests. Are they what you expected?
Compare the “serial fraction” estimated in each test. Are they the same? Why or why not?
Solution
Remember that Gustafson’s Law models the case where the amount of work grows in proportion to the number of processors. In our case, the problem size is the number of pixels and we use a thread for each processor. So to properly evaluate weak scaling in this example you must keep the product of the height and width in direct proportion to the number of threads.
One simple way to do this is to scale only the width while keeping the height constant, or vice versa. This is valid and should give you good results. Or you can carefully choose heights and widths in pairs that give proportional products, like 1000x1000 for one thread, 2000x3000 for 6 threads, and 4000x4000 for 16 threads. Also valid, although perhaps more error-prone.
Below we show a script that scales both height and width by the square root of the thread count, so that their product remains in direct proportion to the thread count and the aspect ratio of the image remains constant. It uses a couple of shell utilities which you may not have seen before,
bcandprintf.#!/bin/bash #SBATCH --cpus-per-task=16 #SBATCH --time=5 #SBATCH --mem-per-cpu=1000M wbase=2000 hbase=2000 for nthreads in $(seq 1 16) do width=$(printf '%.0f' `echo "scale=6;sqrt($nthreads)*$wbase" | bc`) height=$(printf '%.0f' `echo "scale=6;sqrt($nthreads)*$hbase" | bc`) echo "height x width: $height x $width" for replicate in $(seq 1 3) do ./a.out $height $width $nthreads done doneThere are a lot of factors that can affect run time, not all of which are easily controlled in a shared computing environment. For example, other processes running on the same machine as your test might affect the run time.
Scheduling Threads in OpenMP
In the OpenMP module later in this summer school we’ll describe how you can affect how OpenMP divides up parallel work using the ‘schedule’ directive. Here’s a preview:
The schedule refers to the way the work chunks are spread across threads. A static schedule means that it is decided very simply when the loop begins which thread will handle which iterations of the loop. A dynamic schedule means that each thread will work on a few iterations and then take the next chunk which hasn’t been taken by another thread. The latter allows better balancing if the work varies between different iterations, but requires some communication overhead.
Change ‘schedule(dynamic)’ to ‘schedule(static)’ at line 140 in julia_set_openmp.c, recompile the code, and rerun the strong scaling test. Compare test results with dynamic and static scheduling.
Which performs better for this Julia set calculation? Why do you suppose that is?
References:
- Amdahl, Gene M. (1967). AFIPS Conference Proceedings. (30): 483–485. doi: 10.1145/1465482.1465560
- Gustafson, John L. (1988). Communications of the ACM. 31 (5): 532–533. doi: 10.1145/42411.42415
Key Points
An increase of the number of processors usually leads to a decrease in efficiency.
An increase of problem size usually leads to an increase in efficiency.
A parallel problem can often be solved efficiently by increasing the number of processors and the problem size simultaneously. This is called “weak scaling”.
Not every problem is amenable to weak scaling.
Independent Tasks and Job Schedulers
Overview
Teaching: 5 min
Exercises: 5 minQuestions
How to run several independent jobs in parallel?
Objectives
Be able to identify a problem of independent tasks
Use a scheduler like SLURM to submit an array job
Some problems are easy to parallelize. If the sub-tasks don’t need information from each other, or if the sub-tasks could be started in any order (which is more-or-less the same thing), then things are pretty simple.
As an example, there was a researcher who had about 10,000 images of the seafloor collected by a submersible robot, and a single-thread program which quantified the features of one image. (E.g., how many blobs are there? How big are they?) Running the program on a single image took about an hour, so doing the whole data set sequentially would take about 10,000 hours. But there was no reason the program couldn’t be run on different CPU cores or even different computers at the same time.
Computer scientists sometimes call these “embarrassingly parallel” problems. We call them “perfectly parallel” because they can be extremely efficient. A “parameter sweep” is a common example of this type of problem.
Independent tasks in your field?
Can you think of a problem in your field that can be formulated as a set of independent tasks?
We don’t need a fancy parallel programming interface to handle these. We can use a simple task scheduler like GNU Parallel or a dynamic resource manager like Slurm, LSF, PBS, Torque, Grid Engine, etc. Most DRMs support “job arrays”, also called “array jobs” or “task arrays”.
- SLURM: Job Arrays
Here’s an example of a submission script for an array job with the SLURM scheduler.
#!/bin/bash
#SBATCH --array=1-100
#SBATCH --time=0-00:01:00
echo "This is task $SLURM_ARRAY_TASK_ID on $(hostname) at $(date)"
If the above is saved into a script called array_job_submit.sh it can be submitted to the SLURM scheduler with:
$ sbatch array_job_submit.sh
The creation of a Julia set image that we did in the last episode was also nearly perfectly parallel:
- Computing the value of any pixel does not depend on the results for any other pixel.
- The pixels could therefore be computed in any order.
Why not to use a DRM or similar tool?
Given the above, why didn’t we do the Julia set exercise using Slurm or GNU Parallel?
Discussion
Two reasons:
The Julia set problem is not 100% independent: Each sub-task needs to write its result back to the image array, which is a shared piece of memory. It’s not practical to manage access to that piece of shared memory from multiple processes.
There are costs to scheduling, starting, and cleaning up a DRM job or a GNU Parallel task. On Alliance clusters we advise people to estimate that a Slurm job might take as much as a minute of overhead. So the sub-tasks or “work units” should be significantly larger than a minute if you’re going to use Slurm to manage them– an hour or more, preferably.
Key Points
Input and Output
Overview
Teaching: 10 min
Exercises: 5 minQuestions
How is input/output in the HPC clusters organized?
How do I optimize input/output in HPC environment?
Objectives
Sketch the storage structure of a generic, typical cluster
Understand the terms “local disk”, “IOPS”, “SAN”, “metadata”, “parallel I/O”
Cluster storage
Cluster Architecture
Here is the basic architecture of an HPC cluster:
- There is a login node (or maybe more than one).
- There might be a data transfer node (DTN), which is a login node specially designated for doing data transfers.
- There are a lot of compute nodes. The compute nodes may or may not have local disks.
- Most I/O goes to a storage array or SAN (Storage Array Network).
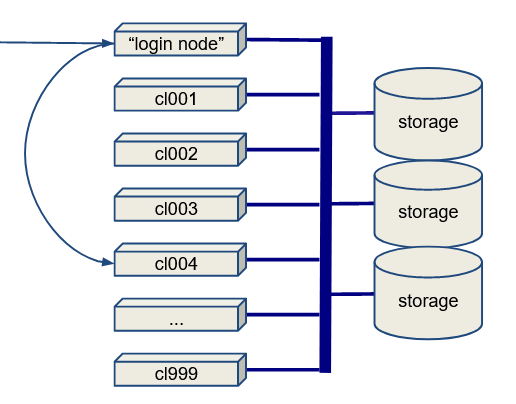
Broadly, you have two choices: You can do I/O to the node-local disk (if there is any), or you can do I/O to the SAN. Local disk suffers little or no contention but is inconvenient.
Local disk
What are the inconveniences of a local disk? What sort of work patterns suffer most from this? What suffers the least? That is, what sort of jobs can use local disk most easily?
Discussion
In a shared computing environment with a DRM running (e.g. Slurm) you cannot know in advance which machine will run your job, so you can’t stage the data needed onto the node in advance. Moving the input data onto the node and the output data off the node must be incorporated into the job script.
If you’re running a distributed-memory job that uses more than one node, then the data has to be distributed and collected from all the nodes, or else all I/O has to be done by a single node— which may become a bottleneck to performance.
Some distributed-memory programs use parallel I/O, which means that different processes may write to a single file at the same time. This is not possible with node-local storage since the nodes don’t share files.
Jobs that use a lot of temporary files which are discarded at the end of the job are very well-suited to node-local storage.
Jobs that make do many small I/O operations (e.g. writing detailed progress information to a log file) are very poorly suited to network storage (SAN), and so are well-suited to node-local storage by comparison.
Mixed use of node and network storage is also possible, e.g. reading data from the SAN but writing results to local disk before staging them back at the end of the calculation, or at infrequent intervals during the calculation.
Local disk performance depends on a lot of things. If you’re interested you can get an idea about Intel data center solid-state drives here.
For that particular disk model, sequential read/write speed is 3200/3200 MB/sec, It is also capable of 654K/220K IOPS (IO operations per second) for 4K random read/write.
Storage Array Network
Most input and output on a cluster goes through the SAN. There are many architectural choices made in constructing SAN.
- What technology? Lustre and GPFS are two important ones, used at ACENET and the Alliance.
- How many servers, and how many disks in each? What disks?
- How many MDS? MDS = MetaData Server. Things like
lsonly require metadata. Exactly what is handled by the MDS (or even if there is one) may depend on the technology chosen (e.g. Lustre, GPFS, …). - What switches connect the storage servers, and how many of them?
- Are things wired together with Ethernet, Infiniband, or something else? What’s the wiring topology?
- Where is there redundancy or failovers, and how much?
This is all the domain of the sysadmins, but what should you as the user do about input/output?
Programming input/output.
If you’re doing parallel computing you have choices about how you do input and output.
- One-file-per-process is reliable, portable and simple, but not great for checkpointing and restarting.
- Many small files on a SAN (or anywhere, really) leads to metadata bottlenecks. Most HPC filesystems assume no more than 1,000 files per directory.
- To have many different processes write to the same file at one time is called “parallel I/O”. MPI supports this via a specification called MPI-IO.
- Parallel I/O makes restarts simpler, but it must be written into the program, it can’t be “added on” trivially.
It also requires support from the underlying file system. (Luster and GPFS support it.) - High-level interfaces like NetCDF and HDF5 are highly recommended
- I/O bottlenecks:
- Disk read-write rate. May be alleviated by striping.
- Network bandwidth.
- Switch capacity.
- Metadata accesses (especially if there is only one MDS)
Measuring I/O rates
- Look into IOR
- Read this 2007 paper, Using IOR to Analyze the I/O performance for HPC Platforms
- Neither of these are things to do right this minute!
The most important part you should know is that the parallel filesystem is optimized for storing large shared files that may be accessible from many computing node. If used to store many small size files, it will perform poorly. As you may have been told in our new-user seminar, we strongly recommend that you do not to generate millions of small size files.
—– Takeaways:
- I/O is complicated. If you want to know what performs best, you’ll have to experiment.
- A few large IO operations are better than many small ones.
- A few large files are usually better than many small files.
- High-level interfaces (e.g. HDF5, netCDF) are your friends, but even they’re not perfect.
Moving data on and off a cluster
- “Internet” cabling varies a lot. 100Mbit/s widespread, 1Gbit becoming common, 10Gbit or more between most CC sites
- Remember, there are 8 bits in a byte. Pay careful attention to whether the number you are reading is bits or bytes!
- CANARIE is the Canadian research network. Check out their Network website
-
Firewalls sometimes the problem - security versus speed
- Filesystem at sending or receiving end often the bottleneck (SAN or simple disk)
- What are the typical disk I/O rates? See above! Highly variable!
Estimating transfer times
- How long to move a gigabyte at 100Mbit/s?
- How long to move a terabyte at 1Gbit/s?
Solution
Remember a byte is 8 bits, a megabyte is 8 megabits, etc.
- 1024 MByte/GByte * 8 bit/Byte / 100 Mbit/sec = 81 sec
- 1024 TByte/GByte * 8 bit/Byte / 1 Gbit/sec = 8192 sec, a little under 2 hours and 20 minutes
Remember these are “theoretical maximums”! There is almost always some other bottleneck or contention that reduces this!
- Restartable downloads (wget? rsync? Globus!)
- checksumming (md5sum) to verify the integrity of large transfers
Key Points
Analyzing Performance Using a Profiler
Overview
Teaching: 30 min
Exercises: 20 minQuestions
How do I decide where to begin optimizing my code?
Objectives
Use a profiler to analyze the runtime behaviour of a program.
Identify areas of the code with potential for optimization and/or parallelization.
Programmers often tend to over-think design and might spend a lot of their time optimizing parts of the code that only contributes a small amount to the total runtime. It is easy to misjudge the runtime behaviour of a program.
What parts of the code to optimize?
To make an informed decision what parts of the code to optimize, one can use a performance analysis tool, or short “profiler”, to analyze the runtime behaviour of a program and measure how much CPU-time is used by each function.
We will analyze an example program, for simple Molecular Dynamics (MD) simulations, with the GNU profiler Gprof. There are different profilers for many different languages available and some of them can display the results graphically. Many Integrated Development Environments (IDEs) also include a profiler. A wide selection of profilers is listed on Wikipedia.
Molecular Dynamics Simulation
The example program performs simple Molecular Dynamics (MD) simulations of particles interacting with a simple harmonic potential of the form:
[v(x) = ( sin ( min ( x, \pi/2 ) ) )^2]
It is a modified version of an MD example written in Fortran 90 by John Burkardt and released under the GNU LGPL license.
Every time step, the MD algorithm essentially calculates the distance, potential energy and force for each pair of particles as well as the kinetic energy for the system. Then it updates the velocities based on the acting forces and updates the coordinates of the particles based on their velocities.
Functions in md_gprof.f90
The MD code md_gprof.f90 has been modified from John Burkardt’s version
by splitting out the computation of the distance, force, potential- and
kinetic energies into separate functions, to make for a more interesting
and instructive example to analyze with a profiler.
| Name of Subroutine | Description |
|---|---|
| MAIN | is the main program for MD. |
| INITIALIZE | initializes the positions, velocities, and accelerations. |
| COMPUTE | computes the forces and energies. |
| CALC_DISTANCE | computes the distance of a pair of particles. |
| CALC_POT | computes the potential energy for a pair of particles. |
| CALC_FORCE | computes the force for a pair of particles. |
| CALC_KIN | computes the kinetic energy for the system. |
| UPDATE | updates positions, velocities and accelerations. |
| R8MAT_UNIFORM_AB | returns a scaled pseudo-random R8MAT. |
| S_TO_I4 | reads an integer value from a string. |
| S_TO_R8 | reads an R8 value from a string. |
| TIMESTAMP | prints the current YMDHMS date as a time stamp. |
Regular invocation:
For the demonstration we are using the example md_gprof.f90.
# Download the source code file:
$ wget https://acenet-arc.github.io/ACENET_Summer_School_General/code/profiling/md_gprof.f90
# Compile with gfortran:
$ gfortran md_gprof.f90 -o md_gprof
# Run program with the following parameters:
# 2 dimensions, 200 particles, 500 steps, time-step: 0.1
$ ./md_gprof 2 200 500 0.1
25 May 2018 4:45:23.786 PM
MD
FORTRAN90 version
A molecular dynamics program.
ND, the spatial dimension, is 2
NP, the number of particles in the simulation is 200
STEP_NUM, the number of time steps, is 500
DT, the size of each time step, is 0.100000
At each step, we report the potential and kinetic energies.
The sum of these energies should be constant.
As an accuracy check, we also print the relative error
in the total energy.
Step Potential Kinetic (P+K-E0)/E0
Energy P Energy K Relative Energy Error
0 19461.9 0.00000 0.00000
50 19823.8 1010.33 0.705112E-01
100 19881.0 1013.88 0.736325E-01
150 19895.1 1012.81 0.743022E-01
200 19899.6 1011.14 0.744472E-01
250 19899.0 1013.06 0.745112E-01
300 19899.1 1015.26 0.746298E-01
350 19900.0 1014.37 0.746316E-01
400 19900.0 1014.86 0.746569E-01
450 19900.0 1014.86 0.746569E-01
500 19900.0 1014.86 0.746569E-01
Elapsed cpu time for main computation:
19.3320 seconds
MD:
Normal end of execution.
25 May 2018 4:45:43.119 PM
Compiling with profiling enabled
To enable profiling with the compilers of the GNU Compiler Collection, we
just need to add the -pg option to the gfortran, gcc or g++ command.
When running the resulting executable, the profiling data will be stored
in the file gmon.out. Invoking gprof will then give us a profile.
By default, gprof looks for a data file named gmon.out but needs to
be given the name of the executable as an argument.
We can also use a larger number of particles and more time steps if we want a longer run time.
# Compile with GFortran with -pg option:
$ gfortran md_gprof.f90 -o md_gprof -pg
$ ./md_gprof 2 500 1000 0.1
# ... skipping over output ...
$ /bin/ls -F
gmon.out md_gprof* md_gprof.f90
# Now the file gmon.out has been created.
# Run gprof to view the output:
$ gprof ./md_gprof | less
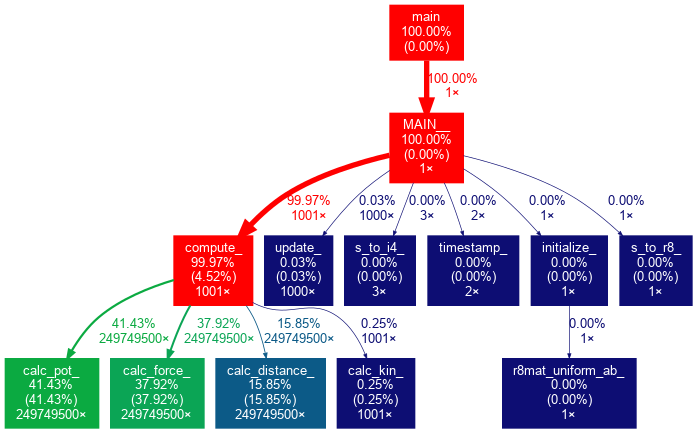
Gprof then displays the profiling information in two tables along with an extensive explanation of their content. We will analyze the tables in the following subsections:
Flat Profile:
Flat profile:
Each sample counts as 0.01 seconds.
% cumulative self self total
time seconds seconds calls s/call s/call name
39.82 1.19 1.19 19939800 0.00 0.00 calc_force_
37.48 2.31 1.12 19939800 0.00 0.00 calc_pot_
17.07 2.82 0.51 19939800 0.00 0.00 calc_distance_
4.68 2.96 0.14 501 0.00 0.01 compute_
1.00 2.99 0.03 501 0.00 0.00 calc_kin_
0.00 2.99 0.00 500 0.00 0.00 update_
0.00 2.99 0.00 3 0.00 0.00 s_to_i4_
0.00 2.99 0.00 2 0.00 0.00 timestamp_
0.00 2.99 0.00 1 0.00 2.99 MAIN__
0.00 2.99 0.00 1 0.00 0.00 initialize_
0.00 2.99 0.00 1 0.00 0.00 r8mat_uniform_ab_
0.00 2.99 0.00 1 0.00 0.00 s_to_r8_
| Column | Description |
|---|---|
| % time | the percentage of the total running time of the program used by this function. |
| cumulative seconds | a running sum of the number of seconds accounted for by this function and those listed above it. |
| self seconds | the number of seconds accounted for by this function alone. This is the major sort for this listing. |
| calls | the number of times this function was invoked, if this function is profiled, else blank. |
| self s/call | the average number of milliseconds spent in this function per call, if this function is profiled, else blank. |
| total s/call | the average number of milliseconds spent in this function and its descendents per call, if this function is profiled, else blank. |
| name | the name of the function. This is the minor sort for this listing. |
In this example, the most time is spent computing the forces and potential energy. Calculating the distance between the particles is at 3rd rank and roughly 2x faster than either of the above.
Call Graph:
The Call Graph table describes the call tree of the program, and was sorted by the total amount of time spent in each function and its children.
Call graph
granularity: each sample hit covers 2 byte(s) for 0.33% of 2.99 seconds
index % time self children called name
0.14 2.85 501/501 MAIN__ [2]
[1] 100.0 0.14 2.85 501 compute_ [1]
1.19 0.00 19939800/19939800 calc_force_ [4]
1.12 0.00 19939800/19939800 calc_pot_ [5]
0.51 0.00 19939800/19939800 calc_distance_ [6]
0.03 0.00 501/501 calc_kin_ [7]
-----------------------------------------------
0.00 2.99 1/1 main [3]
[2] 100.0 0.00 2.99 1 MAIN__ [2]
0.14 2.85 501/501 compute_ [1]
0.00 0.00 500/500 update_ [8]
0.00 0.00 3/3 s_to_i4_ [9]
0.00 0.00 2/2 timestamp_ [10]
0.00 0.00 1/1 s_to_r8_ [13]
0.00 0.00 1/1 initialize_ [11]
-----------------------------------------------
<spontaneous>
[3] 100.0 0.00 2.99 main [3]
0.00 2.99 1/1 MAIN__ [2]
-----------------------------------------------
1.19 0.00 19939800/19939800 compute_ [1]
[4] 39.8 1.19 0.00 19939800 calc_force_ [4]
-----------------------------------------------
1.12 0.00 19939800/19939800 compute_ [1]
[5] 37.5 1.12 0.00 19939800 calc_pot_ [5]
-----------------------------------------------
0.51 0.00 19939800/19939800 compute_ [1]
[6] 17.1 0.51 0.00 19939800 calc_distance_ [6]
-----------------------------------------------
0.03 0.00 501/501 compute_ [1]
[7] 1.0 0.03 0.00 501 calc_kin_ [7]
-----------------------------------------------
0.00 0.00 500/500 MAIN__ [2]
[8] 0.0 0.00 0.00 500 update_ [8]
-----------------------------------------------
0.00 0.00 3/3 MAIN__ [2]
[9] 0.0 0.00 0.00 3 s_to_i4_ [9]
-----------------------------------------------
0.00 0.00 2/2 MAIN__ [2]
[10] 0.0 0.00 0.00 2 timestamp_ [10]
-----------------------------------------------
0.00 0.00 1/1 MAIN__ [2]
[11] 0.0 0.00 0.00 1 initialize_ [11]
0.00 0.00 1/1 r8mat_uniform_ab_ [12]
-----------------------------------------------
0.00 0.00 1/1 initialize_ [11]
[12] 0.0 0.00 0.00 1 r8mat_uniform_ab_ [12]
-----------------------------------------------
0.00 0.00 1/1 MAIN__ [2]
[13] 0.0 0.00 0.00 1 s_to_r8_ [13]
-----------------------------------------------
Each entry in this table consists of several lines. The line with the index number at the left-hand margin lists the current function. The lines above it list the functions that called this function, and the lines below it list the functions this one called.
This line lists:
| Column | Description |
|---|---|
| index | A unique number given to each element of the table. |
| % time | This is the percentage of the `total’ time that was spent in this function and its children. |
| self | This is the total amount of time spent in this function. |
| children | This is the total amount of time propagated into this function by its children. |
| called | This is the number of times the function was called. |
| name | The name of the current function. The index number is printed after it. |
Index by function name:
Index by the function name
[2] MAIN__ [5] calc_pot_ [9] s_to_i4_
[6] calc_distance_ [1] compute_ [13] s_to_r8_
[4] calc_force_ [11] initialize_ [10] timestamp_
[7] calc_kin_ [12] r8mat_uniform_ab_ [8] update_
Plotting the Call Graph
The Call Graph generated by Gprof can be visualized using two tools written in Python: Gprof2Dot and GraphViz.
Install GraphViz and Gprof2Dot
These two packages need to be installed using pip.
On a production cluster you might want to create a
virtual environment
and use the --user option, like this:
$ module load python
$ virtualenv --no-download ~/Profiling_env
$ source ~/Profiling_env/bin/activate
$ pip install graphviz --no-index
$ pip install gprof2dot
On the training cluster the following is sufficient:
$ module load python
$ pip install graphviz gprof2dot
Generate the plot
The graphical representation of the call graph can be created by piping
the output of gprof into gprof2dot and it’s output further into dot
from the GraphViz package. It can be saved in different formats, e.g. PNG
(-Tpng) and under a user-defined filename (argument -o).
If a local X-server is running and X-forwarding is enabled for the current
SSH session, we can use the display command from the ImageMagick tools
to show the image. Otherwise, we can download it and display it with a program
of our choice.
$ gprof ./md_gprof | gprof2dot -n0 -e0 | dot -Tpng -o md_gprof_graph.png
$ display md_gprof_graph.png
The call graph, visualized
Optional exercise
Create different profiles by calling the program with different parameters, e.g.
md_gprof 2 200 500 0.1andmd_gprof 2 500 1000 0.1.What doesn’t change? What does? Does that change your mind about which part of the program you should focus on first?
gprof2dot options
By default
gprof2dotwon’t display nodes and edges below a certain threshold. Because our example has only a small number of subroutines/functions, we have used the-nand-eoptions to set both thresholds to 0%.Gprof2dot has several more options to, e.g. limit the depth of the tree, show only the descendants of a function or only the ancestors of another. Different coloring schemes are available as well.
$ gprof2dot --helpUsage: gprof2dot [options] [file] ... Options: -h, --help show this help message and exit -o FILE, --output=FILE output filename [stdout] -n PERCENTAGE, --node-thres=PERCENTAGE eliminate nodes below this threshold [default: 0.5] -e PERCENTAGE, --edge-thres=PERCENTAGE eliminate edges below this threshold [default: 0.1] -f FORMAT, --format=FORMAT profile format: axe, callgrind, hprof, json, oprofile, perf, prof, pstats, sleepy, sysprof or xperf [default: prof] --total=TOTALMETHOD preferred method of calculating total time: callratios or callstacks (currently affects only perf format) [default: callratios] -c THEME, --colormap=THEME color map: color, pink, gray, bw, or print [default: color] -s, --strip strip function parameters, template parameters, and const modifiers from demangled C++ function names --colour-nodes-by-selftime colour nodes by self time, rather than by total time (sum of self and descendants) -w, --wrap wrap function names --show-samples show function samples -z ROOT, --root=ROOT prune call graph to show only descendants of specified root function -l LEAF, --leaf=LEAF prune call graph to show only ancestors of specified leaf function --depth=DEPTH prune call graph to show only descendants or ancestors until specified depth --skew=THEME_SKEW skew the colorization curve. Values < 1.0 give more variety to lower percentages. Values > 1.0 give less variety to lower percentages -p FILTER_PATHS, --path=FILTER_PATHS Filter all modules not in a specified path
Key Points
Don’t start to parallelize or optimize your code without having used a profiler first.
A programmer can easily spend many hours of work “optimizing” a part of the code which eventually speeds up the program by only a minuscule amount.
When viewing the profiler report, look for areas where the largest amounts of CPU time are spent, working your way down.
Pay special attention to areas that you didn’t expect to be slow.
Thinking in Parallel
Overview
Teaching: 30 min
Exercises: 15 minQuestions
How do I re-think my algorithm in parallel?
Objectives
Take the MD algorithm from the profiling example and think about how this could be implemented in parallel.
Our Goals
- Don’t duplicate work.
- Run as much as possible in parallel.
- Keep all CPU-cores busy at all times.
- Avoid processes/threads having to wait long for data.
What does that mean in terms of our MD algorithm?
Sequential Algorithm
The key subroutine in the sequential MD code, we discovered in the previous
episode, is compute(...). You can find it at line 225 of md_gprof.f90, and
the major loop begins at line 295. Written in Python-ish pseudo-code the
whole program looks something like this:
Pseudo Code
initialize()
step = 0
while step < numSteps:
for ( i = 1; i <= nParticles; i++ ):
for ( j = 1; j <= nParticles; j++ ):
if ( i != j ) :
calculate_distance(i, j)
calculate_potential_energy(i, j)
# Attribute half of the total potential energy of this pair to particle J.
calculate_force(i, j)
# Add particle J's contribution to the force on particle I.
calculate_kinetic_energy()
update_velocities()
update_coordinates()
finalize()
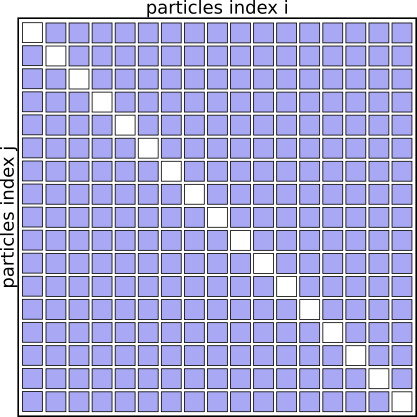
The algorithm basically works through a full matrix of size $N_{particles}^2$, evaluating the distances, potential energies and forces for all pairs (i,j) except for (i==j).
Graphically it looks like this:
Interaction Matrix: pair interactions (i!=j)
Step 1: Improve the sequential code
If you understand the problem domain well enough— and maybe you do, if the pseudo-code makes sense to you already— then you might notice that that interaction matrix will always be symmetrical. We don’t need to evaluate the pairs of particles twice for (i,j) and (j,i), as the contribution to the potential energy for both is always the same, as is the magnitude of the force for this interaction, just the direction will always point towards the other particle.
We can basically speed up the algorithm by a factor of 2 just by eliminating redundant pairs and only evaluating pairs (i<j)
Pseudo Code
initialize()
step = 0
while step < numSteps:
for ( i = 1; i <= nParticles; i++ ):
for ( j = 1; j <= nParticles; j++ ):
if ( i < j ):
calculate_distance(i, j)
calculate_potential_energy(i, j)
# Attribute the full potential energy of this pair to particle J.
calculate_force(i, j)
# Add the force of pair (I,J) to both particles.
calculate_kinetic_energy()
update_velocities()
update_coordinates()
finalize()
Interaction Matrix: pair interactions (i<j)
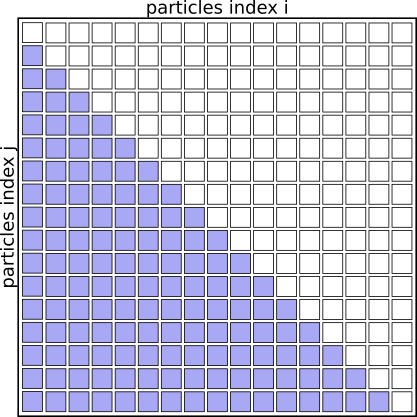
Another way to express the same idea is not to check for i<j at each iteration, but to recode the inner loop to only start counting from i+1, like this:
Pseudo Code
...
for ( i = 1; i <= nParticles; i++ ):
for ( j = i + 1; j <= nParticles; j++ ):
...
At this point, properly we should modify the code, re-compile the program, and
profile it again to find out if our “hot spots” have changed. We’re not going
to take time to do that today. Take my word for it that the compute function
is still the costliest, and that calc_distance and calc_force are still
the major contributors inside of compute.
Step 2: Parallelize the outer loop
It’s not 100% reliable, but a good rule when parallelizing code is to start
with the outermost loop in your “hot spot”. Following that rule in this
example, a good thing to try would be to turn the outer for-loop (over index
i) into a parallel loop. The work for columns i=1,2 will be assigned to
core 1, columns i=3,4 to core 2, and so on. (We will show you
how to actually implement this e.g. using OpenMP in later days of the workshop.)
Pseudo Code
initialize()
step = 0
while step < numSteps:
# Run this loop in parallel:
for ( i = 1; i <= nParticles; i++ ):
for ( j = i + 1; j <= nParticles; j++ ):
calculate_distance(i, j)
calculate_potential_energy(i, j)
# Attribute the full potential energy of this pair to particle J.
calculate_force(i, j)
# Add the force if pair (I,J) to both particles.
gather_forces_and_potential_energies()
# Continue sequentially:
calculate_kinetic_energy()
update_velocities()
update_coordinates()
communicate_new_coordinates_and_velocities()
finalize()
We need to add a step, gather_forces_and_potential_energies because
eventually every particle has to know about every other particle, no
matter which processor the particle is assigned to. That’s not a problem.
Question
There is a different and subtle problem with this scheme. Can you see it in this picture of the interaction matrix?
Interaction Matrix: with simple parallelization
Answer
With this scheme core 1 will be responsible for many more interactions than core 8. So our CPU cores are, on average, idle about 50% of the time, and the whole solution is not available until the most-loaded process finishes.
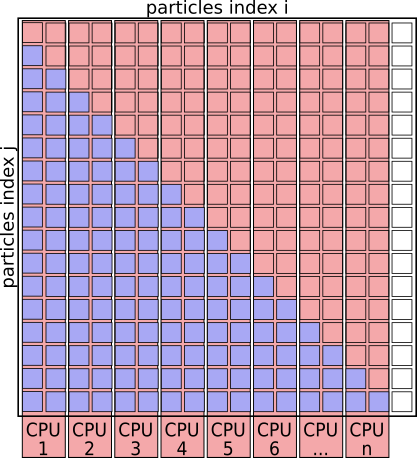
This can be improved by creating a list of particle pairs, and then distribute that list of particle-pairs evenly between cores for evaluation.
Pseudo Code: Using pair-list
initialize()
step = 0
while step < numSteps:
# generate pair-list
pair_list = []
for ( i = 1; i <= nParticles; i++ ):
for ( j = i+1; j <= nParticles; j++ ):
pair_list.append( (i,j) )
# Run this loop in parallel:
for (i, j) in pair_list:
calculate_distance(i, j)
calculate_potential_energy(i, j)
# Attributing the full potential energy of this pair to particle J.
calculate_force(i, j)
# Add the force if pair (I,J) to both particles.
gather_forces_and_potential_energies()
# Continue sequentially:
calculate_kinetic_energy()
update_velocities()
update_coordinates()
communicate_new_coordinates_and_velocities()
finalize()
Using cut-offs
We expect this algorithm to scale as $N_{particles}^2$; that is, the amount of work to be done goes up as the square of the number of particles. The things we’ve done so far haven’t changed that, although we have reduced the amount of work one needs to do per particle pair.
Can we do better? Is there a way to make the amount of work grow more slowly than $N_{particles}^2$?
Once again, knowledge of the problem domain and the algorithm is essential.
At large distances the force between two particles becomes extremely
small, and the contribution of the pair to the potential energy likewise becomes small.
There must be some distance beyond which all forces and
potential-energy contributions are “effectively” zero. Recall that calc_pot
and calc_force were expensive functions in the profile we made of
the code earlier, so we can save time by not calculating these if the
distance we compute is larger than some suitably-chosen $r_{cutoff}$.
Further optimizations can be made by avoiding to compute the distances for all pairs at every step - essentially by keeping neighbor lists and using the fact that particles travel only small distances during a certain number of steps. However, those are beyond the scope of this lesson and are well described in text-books, journal publications and technical manuals.
Spatial- (or Domain-) Decomposition
The scheme we’ve just described, where each particle is assigned to a fixed processor, is called the Force-Decomposition or Particle-Decomposition scheme. When simulating large numbers of particles (~ 105) and using many processors, communicating the updated coordinates, forces, etc., every timestep can become a bottle-neck with this scheme.
To reduce the amount of communication between processors we can partition the simulation box along its axes into smaller domains. The particles are then assigned to processors depending on in which domain they are currently located. This way many pair-interactions will be local within the same domain and therefore handled by the same processor. For pairs of particles that are not located in the same domain, we can use e.g. the “eighth shell” method in which a processor handles those pairs in which the second particle is located only in the positive direction of the dimensions, as illustrated below.
Domain Decomposition using Eight-Shell method
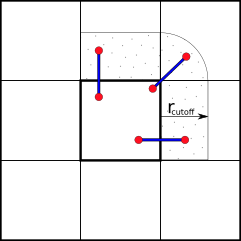
In this way, each domain only needs to communicate with neighboring domains in one direction— as long as the shortest dimension of a domain is larger than the longest cut-off distance.
Domain Decomposition not only applies to Molecular Dynamics (MD) but also to Computational Fluid Dynamics (CFD), Finite Elements methods, Climate- & Weather simulations and many more.
MD Literature:
- Larsson P, Hess B, Lindahl E.;
Algorithm improvements for molecular dynamics simulations.
Wiley Interdisciplinary Reviews: Computational Molecular Science 2011; 1: 93–108. doi:10.1002/wcms.3 - Allen MP, Tildesley DJ; Computer Simulation of Liquids. Second Edition. Oxford University Press; 2017.
- Frenkel D, Smit B; Understanding Molecular Simulation: From Algorithms to Applications. 2nd Edition. Academic Press; 2001.
Load Distribution
Generally speaking, the goal is to distribute the work across the available resources (processors) as evenly as possible, as this will result in the shortest amount of time and avoids some resources being left unused.
Ideal Load: all tasks have the same size
An ideal load distribution might look like this:

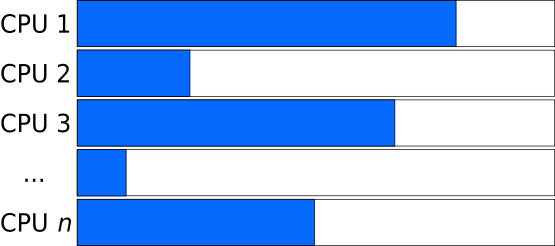
Unbalanced Load: Tasks of different sizes
Whereas if the tasks that are distributed have varying length, the program needs to wait for the slowest task to finish.
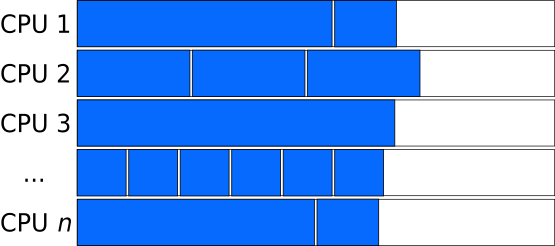
Balanced Load: Combining long and short tasks
If you have many more independent tasks than processors, and if you can estimate the lengths of the tasks easily, then you may be able to combine tasks of different lengths into “chunks” of similar length to balance the load:
Number of chunks a multiple of number of processors
Chunks consisting of many tasks can result in consistent lengths of the chunks, if you are careful (or lucky).
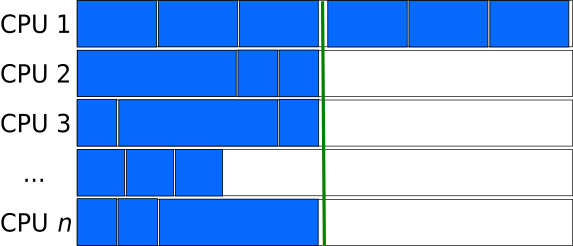
But larger chunks implies a smaller number of chunks. This can cause another inefficiency if the number of chunks is not an integer multiple of the number of processors. In the figure below, for example, the number of chunks is just one larger than the number of processors, so $N-1$ processors are left idle while the $N+1$th chunk is finished up.
Task-queues
Creating a queue (list) of independent tasks which are processed asynchronously can improve the utilization of resources, especially if the tasks can be sorted from longest to shortest.
However, care needs to be taken to avoid a race condition in which two processes take the same task from the task-queue. Having a dedicated manager process to assign the work to the compute processes can eliminate the race-condition, but introduces more overhead (mostly the extra communication required) and can potentially become a bottle-neck when a very large number of processes are involved.
Generally speaking there is a trade-off: Smaller chunks lead to better load balancing, and are hence more efficient. But smaller chunks also usually require more communication (or total storage, or other forms of parallel overhead), and so are less efficient. Assuming you have any choices about chunk size at all, the “best” chunk size for your problem can probably only be determined by trying things.
Key Points
Adapting a sequential code so it will run efficiently parallel needs both planning and experimentation.
It is vital to first understand both the problem and the sequential algorithm.
Shorter independent tasks need more overall communication.
Longer tasks and large variations in task-length can cause resources to be left unused.
Domain Decomposition can be used in many cases to reduce communication by processing short-range interactions locally.
There are many textbooks and publications that describe different parallel algorithms. Try finding existing solutions for similar problems.