Introduction
Overview
Teaching: 15 min
Exercises: 10 minQuestions
Why use MPI?
What is MPI?
How do I run an MPI program?
Objectives
Use
mpirunandsrunRequest resources for MPI from Slurm
Why MPI?
MPI allows utilization of more computers to perform a single task than some of the other parallelization methods we outlined in this workshop (e.g. OpenMP, GPGPU, SIMD). It can also be combined with these other methods. As we have seen there are higher level tools (e.g. Dask, Spark etc. ) which also allow distributed computing. These higher level tools can greatly decrease programming time and provide reasonable parallel speed ups at the expense of the ability to easily understand and control the details of coordination and parallelization.
MPI facilitates communication and coordination among multiple separate machines over a network to perform a single computational task. A problem may be broken up into chunks that each separate machine can operate on and provides a way to communicate results of computations between machines as needed to synchronize and coordinate. One can store a single dataset distributed across the memory of many computers, allowing datasets much larger than the memory of any individual machine to remain in memory while it is operated on.
Below is an example of a fluid dynamics problem solved using this distributed approach with MPI.
This is a model of a star that is radially pulsating with convection occurring near the surface in the hydrogen ionization zone (see sharp change in temperature blue -> red). This simulation is of only a small 2D pie slice section of a full star with periodic boundary conditions at left and right edges. The colour scale indicates the temperature of the gas and the vectors show the convective velocity of the gas. The entire computational grid moves in and out with the radial pulsation of the star. While this 2D simulation admittedly doesn’t push the boundaries of scale it is a good demonstration of how a problem can be solved using MPI.
However this simulation of core convection in a massive star on 1536 cubed grid computed on Compute Canada’s Niagara cluster does push the boundaries of scale. Color shows horizontal velocity. The simulation was conducted by the Computational Stellar Astrophysics group led by Prof Falk Herwig at the University of Victoria.
At the end of the workshop once the necessary building blocks have been presented we will write an MPI parallelized version of a 1D thermal diffusion simulation. While this is admittedly a toy problem many of the important aspects of parallelizing a grid based model will be tackled, such as domain decomposition, guardcells, and periodic boundary conditions. These same problems arose in the radial pulsation simulation and were solved in a similar way to how we will address them in our 1D diffusion simulation. It is also very likely that the core convection simulation had to address similar problems.
What is MPI?
MPI is a standard. MPI stands for Message Passing Interface, a
portable message-passing standard designed by a group of researchers from academia and industry to function on a wide variety of parallel computing architectures. The standard defines the syntax and semantics of a core of library routines useful to a wide range of users writing portable message-passing programs in C, C++, and Fortran. There are several well-tested and efficient implementations of MPI, many of which are open-source or in the public domain.
- The Open MPI implementation is available by default on Compute Canada clusters.
- MPICH and Intel MPI implementations are also available. Use
module spiderimpi/mpichto see module description and details about how to load those modules.
Running multiple copies of a program
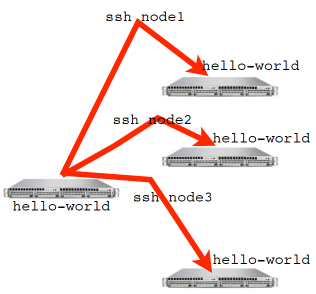
An MPI program, or any program for that matter, can be run with mpirun. mpirun will start N copies of a program. For a mutlinode run mpirun knows the list of nodes your program should run on and ssh’s (or equivalent) to each node and launches the program.
$ ls
mpi-tutorial projects scratch
$ mpirun -np 2 ls
mpi-tutorial projects scratch
mpi-tutorial projects scratch
$ hostname
$ mpirun -np 4 hostname
Multiple ways to launch an MPI program
In addition there is also the commands
mpiexecandsrunwhich can also be used to launch MPI programs.srunis provided by the Slurm scheduler (see slurm srun docs) and provides cluster administrators with better job account than by just runningmpirunormpiexec. Ultimatelysruncalls down to eithermpirunormpiexecto do the actual launching of the MPI program.
What else does mpirun do?
- Assigns an MPI rank to each process
- Sets variables that programs written for MPI recognize so that they know which process they are
Number of Processes
- Number of processes to use is almost always equal to the number of processors
- You can choose different numbers if you want, but think about why you want to!
Interaction with the job scheduler
mpirun behaves a little differently in a job context than outside it.
A cluster scheduler allows you to specify how many processes you want,
and optionally other information like how many nodes to put those
processes on. With Slurm, you can choose the number of process a job has with the option --ntasks <# processes>.
Common slurm options for MPI
The most commonly used slurm options for scheduling MPI jobs are:
--ntasks, specifies the number of process the slurm job has--nodes, specifies the number of nodes the job is spread across--ntasks-per-node, specifies the number of tasks or processes per nodeOften you use either:
--ntasksif you want to merely specify how many process you want but don’t care how they are arranged with respect to compute nodes,or
- you use the combination of
--nodesand--ntasks-per-nodeto specify how many whole nodes and how many process on each node you want. Typically this would be done if your job can make use of all the cores on one or more nodes. For more details on MPI job scheduling see Compute Canada documentation on MPI jobs.
Try this
What happens if you run the following directly on a login node?
$ mpirun -np 2 hostname $ mpirun hostnameHow do you think
mpirundecides on the number of processes if you don’t provide-np?Now get an interactive shell from the job scheduler and try again:
$ salloc --ntasks=4 --time=2:0:0 $ mpirun hostname $ mpirun -np 2 hostname $ mpirun -np 6 hostname
Key Points
mpirunstarts any program
srundoes same asmpiruninside a Slurm job
mpirunandsrunget task count from Slurm context